Введение
Наследственный транстиретиновый амилоидоз (ATTRv) — фатальное системное прогрессирующее заболевание, вызванное мутациями в гене транстиретина (TTR), наследуемое по аутосомно-доминантному типу, преимущественно поражающее периферическую и вегетативную нервную систему и сердце, а также почки, печень, глаза, желудочно-кишечный тракт [1].
Исторически семейный ATTR считался редким эндемическим заболеванием, однако совершенствование диагностики, включая генетические и иммуногистохимические методы, позволило выявить значительно большее число пациентов с наследственным транстиретиновым амилоидозом по всему миру [2]. Знание патомеханизмов, находящихся в основе заболевания, привело к разработке новых и многообещающих препаратов, которые меняют естественное течение семейного ATTR, улучшают качество жизни и выживаемость. Учитывая появление эффективных методов терапии, перед клиницистами стоит задача своевременной диагностики для направления больных на адекватное лечение [3]. Кроме того, прогрессирующий характер ATTRv ставит вопрос о досимптомном тестировании родственников первой линии родства больного пациента для прогностического генетического консультирования носителей мутации в гене TTR и разработки индивидуальных схем диспансерного наблюдения [4].
В статье представлен клинический случай семейного ATTRv-амилоидоза, связанного с выявлением ранее неописанного в литературных источниках варианта нуклеотидной последовательности в гене TTR, ответственного за развитие наследственного транстиретинового амилоидоза.
Клинический случай
Пациент П. с 2018 г. (в возрасте 46 лет) стал отмечать появление одышки при умеренной физической нагрузке, редкие перебои в работе сердца, однако к врачам не обращался. С лета 2020 г. наблюдал ухудшение состояния, появились отеки нижних конечностей с обеих сторон, жалобы на одышку усилились, в связи с чем был госпитализирован по месту жительства с явлениями декомпенсации хронической сердечной недостаточности (ХСН) по большому и малому кругам кровообращения. По данным эхокардиографии (ЭхоКГ) впервые выявлены концентрическая гипертрофия миокарда левого желудочка, расширение предсердий, правого желудочка, фракция выброса (ФВ) — 32%. NT-proBNP — 3296,2 пг\мл. При помощи холтеровского электрокардиографического мониторирования (ХМ-ЭКГ) на фоне синусового ритма регистрировались пароксизмы трепетания, мерцания предсердий, желудочковая экстрасистолия, блокада левой ножки пучка Гиса, преходящая атриовентрикулярная (АВ) блокада I ст. Семейный анамнез по кардиомиопатиям, ХСН, внезапной сердечной смерти не отягощен, у сестры — артериальная гипертензия. В декабре 2020 г. в возрасте 48 лет поступил в отдел заболеваний миокарда и сердечной недостаточности ФГБУ «Национальный медицинский исследовательский центр кардиологии им. акад. Е.И. Чазова» Минздрава России (ФГБУ «НМИЦ кардиологии им. акад. Е.И. Чазова») с направительным диагнозом гипертрофическая кардиомиопатия. При поступлении отмечены явления декомпенсации ХСН по малому и большому кругам кровообращения (отеки нижних конечностей до нижней трети голеней с обеих сторон, по данным рентгенологического исследования органов грудной клетки — венозный застой II ст., УЗ-признаки венозного застоя в системе нижней полой вены (НПВ), свободной жидкости в правой плевральной полости).
Исходя из лабораторных исследований, у пациента при поступлении обращало внимание повышение уровня мозгового натрийуретического пептида до 360,3 пг/мл, увеличение уровня общего билирубина до 46,3 мкмоль/л, гамма-глутамилтранспептидаза — 365,0 Ед/л, по данным общего анализа мочи — повышение уробилиногена до 70 мкмоль/л, в остальном показатели были в пределах нормальных значений.
На ЭКГ — атриовентрикулярная блокада I ст., блокада левой ножки пучка Гиса (рис. 1). Низкий вольтаж QRS в стандартных отведениях и недостаточный прирост зубца R в отведениях V1—V3.

Рис. 1. Электрокардиограмма пациента П.
На ЭхоКГ выявлено выраженное концентрическое утолщение всех стенок левого желудочка (ЛЖ) с преимущественным поражением межжелудочковой перегородки (МЖП), утолщение первичной части межпредсердной перегородки (МПП): толщина МЖП (ТМЖП) составила 3,0—3,2 см, толщина задней стенки ЛЖ (ТЗСЛЖ) — 2,0—2,2 см. Уменьшение объема ЛЖ, дилатация полостей предсердий. Рестриктивный тип нарушения диастолической функции ЛЖ с повышением давления его наполнения (III ст.). Глобальная сократимость ЛЖ снижена диффузно (ФВ ЛЖ 40—42%). Регургитация митрального клапана (МК) II ст., трикуспидального клапана (ТК) II ст. Легочная гипертензия II ст. (посткапиллярная). Выраженная дилатация НПВ со значительным уменьшением коллабирования на вдохе. Небольшое количество жидкости в плевральных полостях с обеих сторон, преимущественно справа. По результатам 3D-ЭхоКГ нельзя было исключить дополнительные включения в миокард ЛЖ, в особенности в МЖП — глыбки амилоида (рис. 2, а).

Рис. 2. Эхокардиограмма больного П.
а — выраженное концентрическое утолщение всех стенок левого желудочка с преимущественным поражением межжелудочковой перегородки, утолщение первичной части межпредсердной перегородки (четырехкамерная позиция); б — технология спекл-трекинг эхокардиограммы (Peak systolic strain). Выраженное снижение деформации миокарда базальных и средних сегментов левого желудочка с относительно сохраненной деформацией миокарда в области верхушки.
Была выполнена ЭхоКГ с использованием метода спекл-трекинга (Speckle Tracking Imaging) с оценкой деформации миокарда [5]. По данным метода спекл-трекинг ЭхоКГ (AFI) отмечалось выраженное снижение деформации миокарда базальных и средних сегментов ЛЖ с относительно сохраненной деформацией миокарда в области верхушки — характерная картина для амилоидоза сердца. Показатель глобальной продольной деформации (Global Longitudinal Strain, GLS) был значительно снижен и составил GLS= –6,5% (N>20%) (рис. 2, б).
На основании полученных данных, была заподозрена амилоидная кардиомиопатия (АК). Анализ периферической крови и суточной мочи на моноклональную секрецию не выявил амилоидогенный клон плазматических клеток, что позволило исключить наиболее частую причину АК — AL-тип амилоидоза.
Пациенту с характерными признаками амилоидоза сердца по данным ЭхоКГ была проведена сцинтиграфия миокарда с остеотропным радиофармпрепаратом (РФП) 99mTc—пирофосфатом для верификации ATTR-амилоидоза. Определялось интенсивное тотальное накопление РФП в миокарде ЛЖ и ПЖ. Соотношение накопления РФП в миокарде относительно накопления в контралатеральной зоне 2.5 (соотношение больше чем 1.5 характерно для ATTR-амилоидоза). Более того, при оценке сцинтиграмм в режиме однофотонной эмиссионной компьютерной томографии (ОЭКТ)/КТ (необходимом для выполнения всем пациентам с подозрением на ATTR АК) визуализировалось интенсивное накопление РФП во всем миокарде левого желудочка, в том числе в проекции папиллярных мышц (признак гипертрофии ЛЖ), Grade III. Сцинтиграфических признаков острого повреждения миокарда не выявлено. Дано заключение, что сцинтиграфическая картина может соответствовать изменениям при ATTR-амилоидозе (рис. 3).

Рис. 3. Сцинтиграмма миокарда с 99mTc—пирофосфатом.
При оценке сцинтиграмм в режиме однофотонного эмиссионного компьютерного томографа/компьютерного томографа визуализируется интенсивное накопление радиофармпрепаратов во всем миокарде левого желудочка, в том числе в проекции папиллярных мышц (признак гипертрофии левого желудочка), Grade III. По данным компьютерной томограммы — плевральный выпот справа до 47 мм.
С целью морфологического уточнения диагноза была проведена биопсия 12 п.к, которая подтвердила наличие депозитов амилоида. Согласно консенсусному алгоритму неинвазивной диагностики амилоидоза сердца (2019), у пациентов с характерными признаками амилоидоза по данным ЭхоКГ/магнитно-резонансной томографии, Grade II/III по данным ОЭКТ и без клональных аномалий диагноз ATTR-амилоидоз может считаться установленным без проведения эндомиокардиальной биопсии миокарда.
Для окончательного суждения о диагнозе был направлен анализ на генетическое тестирование в Центр молекулярной генетики (Москва), где методом прямого секвенирования всей кодирующей последовательности и областей экзон-интронных соединений гена TTR, ответственного за развитие наследственного транстиретинового амилоидоза, был выявлен ранее не описанный в литературных источниках вариант нуклеотидной последовательности NM_000371.4:c.266A>T(NP_000362.1:p.Tyr89Phe, p.Y89F; устаревшее название мутации Tyr69Phe) в гетерозиготном состоянии.
Таким образом, у пациента был диагностирован наследственный ATTR с преимущественным поражением сердца, а также начальными признаками развития периферической полинейропатии (слабость, онемение рук). Функция почек не изменена, протеинурии в моче не отмечалось. Данный диагноз характеризуется высоким риском неблагоприятного прогноза, в связи с чем пациенту по жизненным показаниям был назначен прием тафамидиса 61 мг/сут в качестве патогенетической терапии для улучшения качества жизни, снижения частоты госпитализаций и риска сердечно-сосудистых событий.
При сборе семейного анамнеза выяснилось, что мать пациента погибла в автокатастрофе в возрасте 45 лет, к врачам с жалобами не обращалась. Отец жив, 82 года. У родной сестры в возрасте 53 лет появились жалобы на одышку при умеренной физической нагрузке, редкие эпизоды учащенного сердцебиения (в анамнезе лабильная артериальная гипертензия). Обращалась к кардиологу по месту жительства, где по данным ЭхоКГ была впервые выявлена дилатация обоих предсердий, выраженная гипертрофия левого желудочка (ЗСЛЖ — 1,5 см, ТМЖП — 1,8 см). По данным ХМ-ЭКГ регистрировалась АВ-блокада I ст., преходящая блокада II ст., неполная блокада правой ножки пучка Гиса. Ввиду наличия длительного анамнеза артериальной гипертензии пациентка наблюдалась по месту жительства с диагнозом «гипертоническое сердце». Регулярно медикаментозную терапию не принимала. Ухудшение состояния отметила спустя 2 года в виде прогрессирования одышки (при минимальной физической нагрузке, прохождении 50—100 м), снижения толерантности к физической нагрузке, нарастании отеков нижних конечностей, потери веса на 10 кг. В октябре 2022 г. в возрасте 55 лет была впервые госпитализирована в ФГБУ «НМИЦ кардиологии им. акад. Е.И. Чазова» с явлениями тяжелой декомпенсации сердечной недостаточности по обоим кругам кровообращения: увеличение печени, нижний край определялся на уровне пупка, выраженные периферические отеки, набухание и пульсация шейных вен, выраженная одышка. Также пациентка предъявляла жалобы на онемение пальцев рук и ног. Генез ХСН предположительно рассматривался как амилоидная кардиомиопатия (наследственный транстиретиновый амилоидоз). На ЭКГ — низкий вольтаж QRS в большинстве отведений. Неполная блокада правой ножки пучка Гиса.
По данным экспертной Эхо-КГ выявлялось значительно выраженное утолщение стенок ЛЖ (ТЗСЛЖ=2,4—2,5 см). Полость ЛЖ уменьшена в размерах и объемах, его сократимость относительно удовлетворительная. КДО ЛЖ=56 мл, КСО ЛЖ=23 мл, ФВ 58%. Нарушение диастолической функции миокарда ЛЖ III ст. (по рестриктивному типу — с признаками повышения давления наполнения). Дилатация обоих предсердий, полости ПЖ. Снижение систолической функции ПЖ. Створки клапанного аппарата сердца утолщены, регургитация ТК 3< –IV ст., МК II ст., ЛК I—II ст.; АК I ст. Легочная гипертензия II ст. (посткапиллярная). Расширение ствола легочной артерии и ее ветвей. НПВ расширена, коллабирование на вдохе значительно снижено — высокое центральное венозное давление. Определялось умеренное количество свободной жидкости в плевральных полостях с обеих сторон, в брюшной полости (в проекции печени, селезенки, между петель кишечника) и следовое — в полости перикарда. 2D- и 3D-ЭхоКГ картина соответствовала выраженной степени амилоидного поражения сердца: с наличием мелких гиперэхогенных включений в миокард с вовлечением клапанного аппарата сердца.
По данным технологии спекл-трекинг ЭхоКГ отмечалось значительное снижение деформации базальных и средних с относительно сохранной деформацией апикальных сегментов ЛЖ, GLS= –5,6% (N>20%); картина характерная для амилоидоза сердца. При сцинтиграфии миокарда с 99mTc-пирофосфатом от 16.12.20: картина может соответствовать изменениям при ATTR-амилоидозе (Grade III).
Также пациентка консультирована неврологом, выявлен полинейропатический синдром. При осмотре офтальмологом окулярных нарушений выявлено не было. В отличие от пробанда, у пациентки отмечалась протеинурия по данным ОАМ (0,69 г/л) с развитием хронической болезни почек C3б стадии (скорость клубочковой фильтрации по формуле CKD-EPI: 43 мл/мин/1,73м2).
Семейный ATTR-амилоидоз наследуется по аутосомно-доминантному типу. Каждый ребенок больного человека (гетерозиготный по одному патогенному варианту TTR) имеет 50% шанс унаследовать вариант TTR (рис. 4).
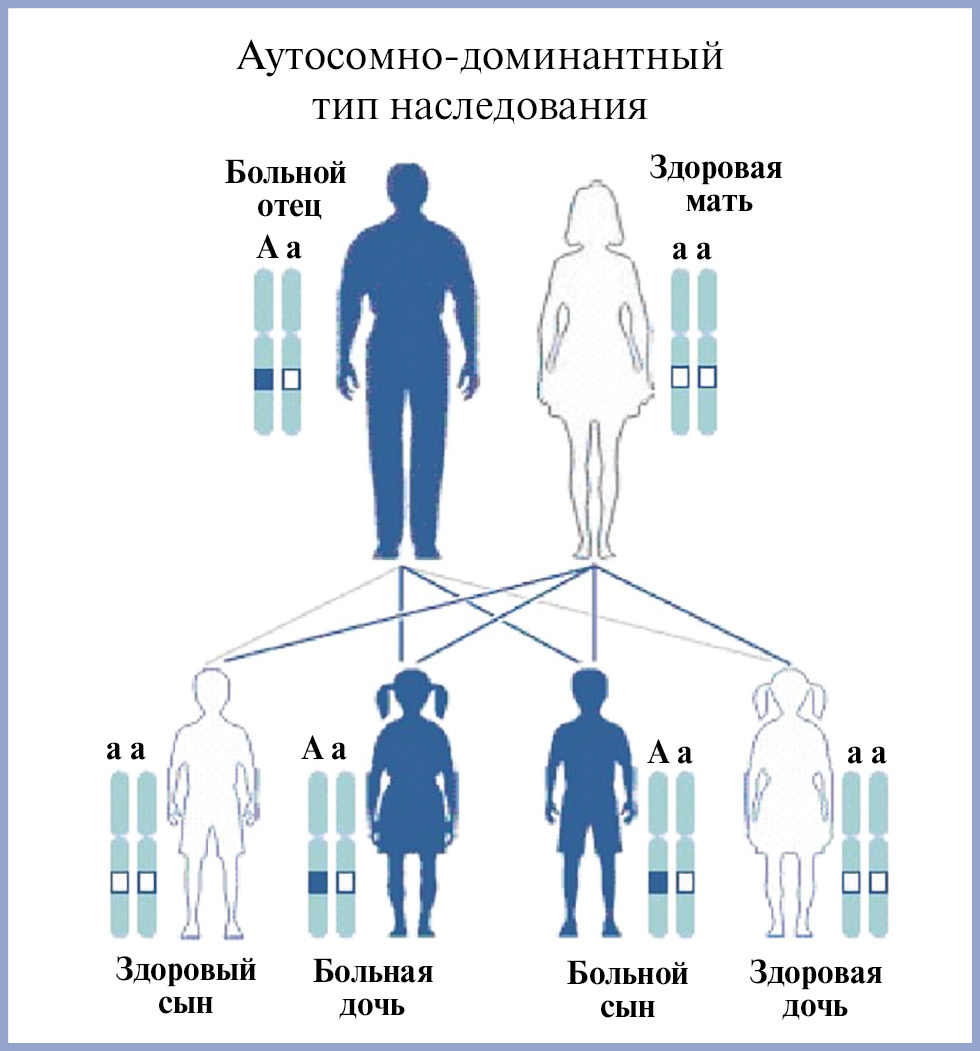
Рис. 4. Схема аутосомно-доминантного типа наследования.
Был проведен генетический скрининг ближайших родственников пробанда для выявления носителей мутации в гене TTR. Секвенирование гена TTR показало, что оба ребенка пациента от разных браков не являются носителями мутации гена TTR. По данным генетического тестирования единственной родной сестры пробанда также выявлен ранее не описанный вариант p.Tyr89Phe (Tyr69Phe) в гетерозиготном состоянии в гене TTR. Кроме того, бессимптомным носителем этой же мутации оказался единственный сын сестры пробанда (рис. 5).

Рис. 5. Семейное древо с вариантом мутации p.Tyr89Phe (Tyr69Phe) в гетерозиготном состоянии в гене TTR. Стрелка указывает пробанда.
Обсуждение
Возраст манифестации, преимущественный фенотип и прогноз зачастую определяются генетическим вариантом ATTRv. Ген TTR, ответственный за развитие наследственного транстиретинового амилоидоза, расположен в хромосоме 18 и включает в себя 4 экзона. На сегодняшний день по данным базы HGMD (Human Gene Mutation Database) в гене TTR описано 162 патогенных варианта, большинство из которых являются патогенными и связаны с различными фенотипами заболевания [6]. Наиболее частым является вариант Val30Met (p.Val50Met). Мутация Val30Met ответственна за высокую распространенность ATTRv в эндемичных районах, включая Португалию, Швецию. Частота носительства заметно различается в зависимости от расового происхождения, что подчеркивается частым появлением мутации ATTRv Val122Ile (p.Val142Ile) в афроамериканском, западноафриканском и латиноамериканском населении [7]. В России по данным молекулярно-генетического анализа 22 неродственных пробандов с ATTR из различных регионов РФ, проведенного в лаборатории ДНК-диагностики ФГБНУ «Медико-генетический научный центр им. акад. Н.П. Бочкова», установлено, что в 59% (13 больных) случаев причиной болезни явилась мутация Val30Met (аналогично большинству европейских стран); по 2 раза встретились вариант Phe33Leu (9%), ранее описанный у пациентов с польскими корнями, и вариант Ile107Val (9%); однократно выявлены патогенные варианты: Ser23Asn, Ala25Thr, Ala81Val, Glu92Lys, Thr40Asn [8].
Большинство мутаций гена TTR, в том числе наиболее частый вариант Val30Met, вызывают развитие периферической сенсомоторной и вегетативной полинейропатии. Основными клиническими проявлениями вегетативной нейропатии являются ортостатическая гипотензия, запоры, чередующиеся с диареей, тошнота и рвота, эректильная дисфункция, ангидроз, задержка или недержание мочи [9]. Однако известен ряд мутаций, ассоциированный с развитием амилоидной кардиомиопатии, при которых периферическая и вегетативная нейропатии отсутствуют или слабо выражены: p.Asp38Asn, p.Val40Ile, p.Pro44Ser, p.Ala65Thr, p.Ala65Ser, p.His76Arg, p.Gly77Arg, p.Ile88Leu, p.Ala101Thr, p.Ala101Val, p.His108Arg, p.Glu112Lys, p.Arg123Ser, p.Leu131Met и p.Val142Ile [10].
Генетическое тестирование рекомендовано для предупреждения родственников о возможных рисках развития наследственного транстиретинового амилоидоза. Родственников пациентов с ATTRv следует рассматривать как возможных носителей семейной мутации и необходимо направлять к экспертной междисциплинарной группе [11]. Хотя все родственники взрослых пациентов могут пройти генетическое тестирование на ATTRv, наибольшим приоритетом обладает тестирование братьев и сестер ввиду близкого расположения к прогнозируемому возрасту начала заболевания и высокой вероятности развития ATTRv в ближайшем будущем [12].
В обследованной нами семье был выявлен ранее не описанный в литературных источниках вариант p.Tyr89Phe (Tyr69Phe) в гене TTR. Проведено сравнение клинических проявлений заболевания и данных диагностических методов исследования амилоидоза членов семьи. В обоих случаях имело место преимущественное развитие амилоидной кардиомиопатии со стремительным прогрессированием от начала появления первых симптомов до развития тяжелой сердечной недостаточности (менее 2 лет). Кардиологические нарушения у пациентов проявлялись нарушениями ритма и проводимости, утолщением стенок желудочков при сохранной фракции выброса и отсутствием дилатации ЛЖ. Кроме того, были выявлены поражения нервной системы, печени и почек, однако наиболее существенным являлось развитие амилоидной кардиомиопатии.
Таблица 1. Сравнение клинических проявлений заболевания
| Признак | Пробанд | Сестра пробанда |
| Возраст манифестации заболевания | 46 лет | 53 лет |
| Возраст постановки диагноза | 48 лет | 55 лет |
| Кардиомиопатия | + | + |
| Поражения нервной системы | + | + |
| Поражения мышц | — | — |
| Признаки поражения ЖКТ | — | — |
| Поражение печени | + | + |
| Признаки поражения глаз | — | — |
| Нефропатия | — | + |
Примечание. ЖКТ — желудочно-кишечный тракт.
Таблица 2. Сравнение данных диагностических методов исследования
| Данные диагностики | Пробанд | Сестра пробанда |
| Показатели ЭхоКГ |
| Аорта (см) | 3,2 | 2,7 |
| ЛП (см) | 5,0 | 5,2 |
| Объем ЛП (мл) | 98 | 85 |
| Индекс объема ЛП (мл/м2) | 46,7 | 45,5 |
| КДР ЛЖ/КДО ЛЖ (см/мл) | 4,6/119 | 3,5/56 |
| КСР ЛЖ/КСО ЛЖ (см/мл) | 3,7/71 | 2,4/23 |
| Индекс КДО ЛЖ (мл/м2) | 56,7 | 29,9 |
| Индекс КСО ЛЖ (мл/м2) | 33,8 | 12,3 |
| ФВ ЛЖ (%) | 40—42 | 58 |
| ТМЖП (см) | 3,0—3,2 | 1,9 |
| ТЗСЛЖ (см) | 2,0—2,2 | 2,4—2,5 |
| Площадь ПП (см2) | 25 | 27 |
| Правый желудочек (см) | 2,9 | 3,5 |
| Нижняя полая вена (см) | 3,9/2,9 коллабирует <50% | 3,0/2,5 коллабирует |
| СДЛА (мм рт.ст.) | 50 | 52—55 |
| Недостаточность МК (ст.) | 2 | 2 |
| Недостаточность ТК (ст.) | 2 | 3 |
| Показатели диастолической функции ЛЖ по ТМД (см/с): | | |
| Em l | 5 | 4 |
| Em s | 3 | 3—4 |
| E/Em | 21 | 30 |
| Показатель деформации миокарда ЛЖ GLS (%) | —6,5 | –5,6 |
| Сцинтиграфия миокарда с 99mTc—пирофосфатом | Grade III | Grade III |
Примечание. ЭхоКГ — эхокардиография; ЛП — левое предсердие; КДР ЛЖ — конечно-диастолический размер левого желудочка; КДО ЛЖ — конечно-диастолический объем левого желудочка; КСР ЛЖ — конечно-систолический размер левого желудочка; КСО ЛЖ — конечно-систолический объем левого желудочка; ФВ ЛЖ — фракция выброса левого желудочка; ТМЖП — толщина межжелудочковой перегородки; ТЗСЛЖ — толщина задней стенки левого желудочка; ПП — правое предсердие; СДЛА — систолическое давление в легочной артерии; МК — митральный клапан; ТК — трикуспидальный клапан; GLS — global longitudinal strain, глобальная продольная деформация.
Необходима разработка протокола генетического тестирования ATTRv, согласно которому в процессе должны участвовать специалисты, обладающие опытом в области генетического консультирования, обеспечения точной интерпретации молекулярных результатов, а также в лечении и последующем наблюдении. Из-за возможного психологического воздействия результатов генетического тестирования в команде также должен быть психолог с опытом генетического консультирования [13].
Несмотря на аутосомно-доминантный тип наследования, заболевание проявляется не в каждом поколении. Выявление мутации у родственников не дает оснований ставить диагноз ATTR-амилоидоза и не гарантирует развитие заболевания в будущем. Однако пациентам с положительным результатом теста необходим ежегодный мониторинг за 10 лет до прогнозируемого возраста начала развития кардиологических и неврологических проявлений заболевания [14]. Определение прогнозируемого возраста основано на конкретной мутации, ее типичном возрасте начала проявления и возрасте дебюта заболевания у членов семьи. Таким образом, необходимо привлечение междисциплинарной команды для ежегодного мониторинга родственников первой линии родства с мутацией в гене TTR (невролога, кардиолога, гастроэнтеролога, офтальмолога) для выявления ранних признаков заболевания и назначения своевременного специфического лечения [15].
Выводы
Данный клинический случай подчеркивает важность проведения генетического тестирования родственников первой линии родства для предупреждения о возможных рисках развития наследственного транстиретинового амилоидоза. Необходима разработка протокола генетического тестирования ATTRv с привлечением междисциплинарной команды специалистов для ежегодного мониторинга родственников первой линии родства с выявленной мутацией в гене TTR для обнаружения ранних симптомов развития амилоидной кардиомиопатии и амилоидной нейропатии и назначения своевременного специфического лечения.
Авторы заявляют об отсутствии конфликта интересов.





